Introduction
Application of soft computing becomes relevant for solving some Bioinformatics and molecular biology problems. Protein classification leads to identification and proper functional assignment of uncharacterized proteins with a final goal towards finding homologies and drug discovery. Again, structure based ligand design is one of the crucial steps in rational drug discovery, where a small molecule is designed by targeting the structure and biochemical properties of the target. The application of soft computing offers an on promising approach to achieve efficient and reliable heuristic solution. On the other side the continuous development of high quality biotechnology, e.g. micro-array techniques and mass spectrometry, which provide complex patterns for the direct characterization of cell processes, offers further promising opportunities for advanced research in bioinformatics. So One important sub-discipline within bioinformatics involves the development of new algorithms and models to extract new, and potentially useful information from various types of biological data including DNA(nucleotide sequences) and proteins (amino acid sequences). Analysis of these macromolecules is performed both structurally and functionally using the major components of soft computing like Fuzzy Sets (FS), Artificial Neural Networks (ANN), Evolutionary Algorithms (EAs) (including genetic algorithms (GAs), genetic programming (GP), evolutionary strategies (ES)), Support Vector Machines (SVM), Wavelets, Rough Sets (RS), Simulated Annealing (SA), Swarm Optimization (SO), Memetic Algorithms (MA), Ant Colony Optimization (ACO) and Tabu Search (TS).
Need for Soft Computing techniques in Bioinformatics
The different tasks involved in the analysis of biological data include Sequence alignment, genomics, proteomics,DNA and protein structure Prediction, gene/promoter identification, phylogenetic analysis, analysis of Gene expression data, protein Folding, docking and molecule and Drug design. Data analysis tools used earlier in bioinformatics were mainly based on statistical techniques like regression and estimation. Soft computing in bioinformatics can be used in handling large, complex, inherently uncertain, data sets in biology in a robust and computationally efficient manner thus fuzzy sets (soft computing technique) can be used as a natural framework for analyzing them. Most of the bioinformatic tasks involve search and optimization of different criteria (like energy, alignment score, overlap strength), while requiring robust, fast and close approximate solutions. Evolutionary and other soft computing search algorithms like TS, SA, ACO, PSO etc. provide powerful searching methods to explore huge and multi-model solution spaces.
In molecular biology research, new data and concepts are generated every day, and those new data and concepts update or replace the old ones. Soft computing can be easily adapted to a changing environment. This benefits system designers, as they do not need to re-design systems whenever the environment changes. Moreover, since many of the problems involve multiple conflicting objectives, application of soft computing multi-objective optimization algorithms like multi-objective genetic algorithms appears to be natural and appropriate. Soft computing techniques, either individually or in a hybridized manner, can be used for analyzing biological data in order to extract more and more meaningful information and insights from them.
Characteristics of the methods and algorithms reported here include the use of domain-specific knowledge for reducing the search space, dealing with uncertainty, partial truth and imprecision, efficient linear and/or sub-linear scalability, incremental approaches to knowledge discovery, and increased level and intelligence of interactivity with human experts and decision makers.
Ant Colony Optimization (ACO) Relevance of ACO in bioinformatics
Ant Colony Optimization (ACO) is a population- based, general search technique for the solution of difficult combinatorial problems which is inspired by the pheromone trail laying behavior of real ant colonies. More specifically, an ant is a simple computational agent, which iteratively constructs a solution for the instance to solve. Partial problem solutions are seen as states. Identification of small subsets of highly predictive and biologically relevant genes in bioinformatics is a tedious task. The ACA(Ant colony Algorithm) is an optimization algorithm capable of incorporating prior information, allowing it to search the sample space more efficiently when applied to several high-dimensional data sets, the ACA can identify small subsets of highly predictive and biologically relevant genes without the need for extensive preselecting of features(KR Robbins et al).Segmentation of Brain MR Images Using an Ant Colony Optimization , this is a relatively new meta-heuristic algorithm and a successful paradigm of all the algorithms which take advantage of the insect’s behavior(Myung-Eun Lee et al). Ant colony algorithm can be used to, the well known bioinformatics problem of aligning several protein sequences (Jonathan Moss et al). Ant colony optimization (ACO) is a promising approach to the problem of interacting DNA sequence variations to effectively explore interactions in these datasets to identify combinations of variations which are predictive of common human diseases (Casey S. Greene et al). Flexible ant colony (FAC) algorithm can be used for solving protein folding problems (PFPs) based on the hydrophobic-polar (HP) square lattice model (Xiao-Min Hu et al).ACO can provide a better genotyping strategy, when compared to A-1, with different pedigree sizes and structures (M. L. Spangler et al).
Support Vector Machines
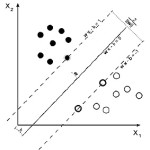
A Support Vector Machine (SVM) performs classification by constructing an N-dimensional hyper plane that optimally separates the data into two categories. SVM models are closely related to neural networks. In fact, a SVM model using a sigmoid kernel function is equivalent to a two-layer, perceptron neural network. Using a kernel function, SVM’s are an alternative training method for polynomial, radial basis function and multi-layer perceptron classifiers in which the weights of the network are found by solving a quadratic programming problem with linear constraints, rather than by solving a non-convex, unconstrained minimization problem as in standard neural network.
Formalization
We are given some training data, a set of points of the form
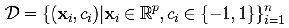
where the ci is either 1 or “1, indicating the class to which the point belongs. Each is a p-dimensional real vector. We want to find the maximum-margin hyper plane which divides the points having ci = 1 from those having ci = -1. Any hyper plane can be written as the set of points satisfying
Maximum-margin hyper plane and margins for a SVM trained with samples from two classes. Samples on the margin are called the support vectors.
W.x-b=0,
Where .denotes the dot product. The vector W is a normal vector: it is perpendicular to the hyper plane.
The parameter determines the offset of the hyper plane from the origin along the normal vector.
We want to choose the and b to maximize the margin, or distance between the parallel hyper planes that are as far apart as possible while still separating the data. These hyper planes can be described by the equations W. X – b = 1 and Note that if the training data are linearly separable, we can select the two hyper planes of the margin in a way that, there are no points between them and then try to maximize their distance. By using geometry, we find the distance between these two hyper planes is , so we want to minimize. As we also have to prevent data points falling into the margin, we add the following constraint: for each i either of the first class or of the second. This can be rewritten as:
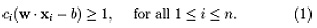
We can put this together to get the optimization problem: Minimize (in W, b)
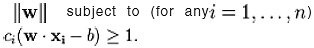
Relevance of SVM in Bioinformatics
Classification problems are very common (structure, function, localization prediction; analysis of microarray data…….)
Small training sets in high dimension is common
An extension of SVM to non-vector objects (strings, graphs) is natural.
Novelty detection techniques might be a promising way of dealing with high-dimensional classification problems in Bioinformatics.
Support vector machine approach can be used to detect novel classes in Bioinformatics databases.
Determining the colour selectivity of visual areas is a major task in Bioinformatics. Support vector machines can be used to determine the color selectivity of visual areas (Dr. Laura Parkes et al).
Biologists use microarrays to study gene expression patterns in a wide range of medical and scientific applications .Support Vector Machine (SVM) is a powerful machine learning technique that can be used in a variety of data mining applications, including the analysis of DNA microarrays (R. Morelli et al).
Applications of SVMS in Bioinformatics
Classification of real and pseudo micro RNA precursors using features and support vector machine (Chenghai Xue et al,2005)
NOXclass: prediction of protein-protein local structure-sequence interaction types (Hongbo Zhu et al, 2006).
Improved prediction of protein–protein binding sites using a support vector machines approach (J. R. Bradford et al, 2005).
Understanding protein dispensability through machine-learning analysis of high-throughput data (Yu Chen et al, 2005).
Accurate identification of alternatively spliced exons using support vector machine (Gideon Dror et al, 2005).
Support vector machines for separation of mixed plant–pathogen EST collections based on codon usage (Caroline C et al, 2005).
Implicit motif distribution based hybrid computational kernel for sequence classification (Volkan Atalay et al, 2005).
Human pol II promoter prediction: time series descriptors and machine learning (Rajeev Gangal et al, 2005).
A global approach to the diagnosis of leukemia using gene expression profiling (Torsten Haferlach et al, 2005).
Concept-based annotation of enzyme classes (Oliver Hofmann et al).
Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information (Lei Bao et al).
Non-additivity in protein–DNA binding (R. A. O’Flanagan et al).
Improved method for predicting _ -turn using support vector machine (Qidong Zhang et al).
An evolution based classifier for prediction of protein interfaces without using protein structures (I. Reš, et al).
Auto Motif server: prediction of single residue post-translational modifications in proteins (Dariusz Plewczynski et al).
Advantages of SVMs in Bioinformatics
Support vector Machines (SVMs) can be employed for approximating the dynamic behaviors of the systems under investigation (Physica D, 2006).
Support vector machines can effectively be used to model the complex relationship between different soil parameter and the liquefaction potential. support vector machines required few user-defined parameters and provide better performance in comparison to neural network approach (Goh ATC,1994)
Support Vector Machines can be used in Geo and Environmental Sciences(M Kanevskiet al,2000)
SVM for Protein Fold and Remote Homology Detection (Huzefa Rangwala et al,2005)
DATA Classification using SSVM (O. L. Mangasarian et al)
Identification of alternative exons using SVM (Dror G et al, 2005)
Support Vector Machines For Texture Classification(Kwang In Kim et al, 2002)
A Comparison of the Performance of Artificial Neural Networks and Support Vector Machines for the Prediction of Traffic Speed and Travel Time (V. Kecman et al).
Support vector classifiers were applied to the recognition of isolated handwritten digits optically scanned (B. Boser et al).
Tabu Search
Tabu search is a metaheuristic algorithm that can be used for solving combinatorial optimization problems, such as the Travelling Salesman Problem (TSP). Tabu search uses a local or neighborhood search procedure to iteratively move from a solution x to a solution x’ in the neighborhood of x, until some stopping criterion has been satisfied. To explore regions of the search space that would be left unexplored by the local search procedure, tabu search modifies the neighborhood structure of each solution as the search progresses. The solutions admitted to N * (x), the new neighborhood, are determined through the use of memory structures. The search then progresses by iteratively moving from a solution x to a solution x’ in N * (x). Perhaps the most important type of memory structure used to determine the solutions admitted to N * (x) is the tabu list. In its simplest form, a tabu list is a short-term memory which contains the solutions that have been visited in the recent past (less than n iterations ago, where n is the number of previous solutions to be stored(n is also called the tabu tenure)). Tabu search excludes solutions in the tabu list from N * (x).
Applications of Tabu Search in Bioinformatics
Application of Tabu search strategy for finding low energy structure of protein is competitive as compared with other methods and due to its low computation time, it can be used as a complementary tool for an analysis of the three-dimensional protein structure (J.B³a¿ewicz et al)
The adaptive memory features of tabu search are implemented to align multiple sequences (Tariq Riaz et al).
Tabu search algorithm is used as a tool for automating nuclear overhauls effect (NOE) pathway, which determines RNA tertiary structure (Jacek Blazewicz et al).
Tabu search algorithm is used for finding a most parsimonious phylogeny tree. Percentage of search space needed to find the best solution for the algorithm decreased rapidly as the number increased (Yu-Min Lin et al).
Tabu Search has been applied for protein structure prediction (Martin Paluszewski et al).
Advantages of Tabu Search in Bioinformatics
The incorporation of tabu search (TS) as a local improvement procedure enables the algorithm Hybrid particle swarm optimization TS to overleap local optima and show satisfactory performance (Qi Shen et al).Tabu Search has been applied to study the reconstruction of a protein’s Cá trace solely from structure-derived HSE information. To discretize the conformational space, lattice models with various complexity is been used (Martin Paluszewski et al).
Conclusion
Bioinformatics is a developing interdisciplinary science. The involvement of other sciences (such as computer science, maths, and physics) holds great promise. The recent research is mainly focused in the biological and health sciences. Computer science departments planning to diversify their offerings can thus only gain through early entry into bioinformatics. The ultimate goal of the field is to enable the discovery of new biological insights as well as to create a global perspective from which unifying principles in biology can be derived. With minimum cost and resources, computer science graduates can excel in their employment. Computer scientists can benefit a lot by collaborating with biologists for new massive inventions.
References
- K. R. Robbins {dagger}, W. Zhang and J. K. Bertrand” The ant colony algorithm for feature selection in high-dimension gene expression data for disease classification”.
- Myung-Eun Lee ,Soo-Hyung Kim, Wan-Hyun Cho, Soon-Young Park ,Jun-Sik Lim “Segmentation of Brain MR Images Using an Ant Colony Optimization Algorithm”.
- Jonathan Moss and Colin G. Johnson.” Anant colony algorithm for multiple sequence alignment in bioinformatics”.
- Casey S. Greene, Bill C. White, and Jason Moore” Ant Colony Optimization for Genome-Wide Genetic Analysis”.
- Xiao-Min Hu, Jun Zhang and Yun Li” Flexible Protein Folding by Ant Colony Optimization”.
- M. L. Spangler, K. R. Robbins, J. K. Bertrand, MacNeil and R. Rekaya “Ant colony optimization as a method for strategic genotype sampling”.
- R. Morelli” Using a Support Vector Machine to Analyze a DNA Microarray”.
- Chenghai Xue, Fei Li, Tao He, Guo-Ping Liu, Yanda Li, Xuegong Zhang. “Classification of real and pseudo microRNA precursors using features and support vector machine”, BMC Bioinformatics,6:310, 2005
- Hongbo Zhu, Francisco S Domingues, Ingolf Sommer, Thomas Lengauer. “NOX class: prediction of protein-proteinlocal structure-sequence interaction types”,BMC Bioinformatics,7:27, 2006
- J. R. Bradford,d David R. Westhead.”Improved prediction of protein– protein binding sites using a support vector machines approach”, BIOINFORMATICS, 21, 8, 1487–1494, 2005.
- Yu Chen, Dong Xu. “Understanding protein dispensability through machine-learning analysis of high-throughput data”, BIOINFORMATICS, 21, 5, 575–581, 2005.
- Gideon Dror, Rotem Sorek, Ron Shamir. “Accurate identification of alternatively spliced exonsusing support vector machine”, BIOINFORMATICS,21, 7, 897–901, 2005.
- Caroline C. Friedel, Katharina H. V. Hahn, Selina Sommer, Stephen Rudd3, Hans W. Mewes,Igor V. Tetko.” Support vector machines for separation of mixed plant– pathogen EST collections based on codon usage”, BIOINFORMATICS, 21, 8, 383– 1388, 2005
- Volkan Atalay, Rengul Cetin-Atalay. “Implicit motif distribution based hybrid computational kernel for sequence classification”, BIOINFORMATICS,21, 8, 1429–1436, 2005
- Rajeev Gangal, Pankaj Sharma.” Human pol II promoter prediction: time series descriptors and machine learning”, Nucleic Acids Research,33, 4, 1332–1336, 2005
- Torsten Haferlach, Alexander Kohlmann, Susanne Schnittger, Martin Dugas, Wolfgang Hiddemann, Wolfgang Kern, Claudia Schoch. “A global approach to the diagnosis of leukemia using gene expression profiling”, Blood First Edition Paper, prepublished onlineMay 5, 2005.
- Oliver Hofmann, Dietmar Schomburg.”Concept-based annotation of enzyme classes”.
- Lei Bao, Yan Cui. “Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information”
- R. A. O’Flanagan, G. Paillard, R. Lavery, A. M. Sengupta. “Non-additivity in protein–DNA binding”.
- Qidong Zhang, Sukjoon Yoon, William J. Welsh. “Improved method for predicting _ – turn using support vector machine”.
- I. Reš, I. Mihalek, O. Lichtarge.” An evolution based classifier for prediction of protein interfaces without using protein structures”.
- Dariusz Plewczynski, Adrian Tkacz1, Lucjan Stanis³aw Wyrwicz, Leszek Rychlewski. “Auto Motif server: prediction of single residue post-translational modifications in proteins”.
- “Dynamic Reconstruction of Chaotic Systems from Inter-spike Intervals Using Least Squares Support Vector Machines” Physica D, Vol. 216, pp. 282-293, 2006)
- Goh ATC. Seismic Liquefaction Potential Assessed by Neural Networks. Journal of Geotechnical Engineering 1994; 120(9): 1467-1480.
- M Kanevski, N Gilardi, E Mayoraz, M Maignan. Spatial Data Classification with Support Vector Machines. Geostat 2000 congress. South Africa, April 2000.
- Huzefa Rangwala, George Karypis.” Profile based direct kernels for remote homology detection and fold recognition”, Bioinformatics, 2005.
- O. L. Mangasarian,” A Smooth Support Vector machine for classification”.
- Dror G., Sorek R. and Shamir S. “Accurate identification of alternatively spliced exons using Support Vector Machine”, Bioinformatics. 2005 Apr 1; 21(7):897-901.
- Kwang In Kim, Keechul Jung, Se Hyun Park, and Hang Joon Kim,” Support Vector Machines for Texture Classification”, IEEE Transactions On Pattern Analysis And Machine Intelligence, Vol. 24, No. 11, November 2002.
- V. Kecman, “Learning and Soft Computing: Support Vector Machines, Neural Networks, And Fuzzy Logic Models”, The MIT press, Cambridge, Massachusetts, London, England.
- B. Boser, I. Guyon, V. Vapnik. “A training algorithm for optimal margin classifiers”.
- J.B³a¿ewicz, P.£ukasiak, M.Mi³ostan.” Application of Tabu search strategy for finding low energy structure of protein”.
- Tariq Riaz, Yi Wang and Kuo-bin Li.” Multiple sequence alignment using tabu search’.
- Jacek Blazewicz, Marta Szachniuk and Adam Wojtowicz.” RNA tertiary structure determination: NOE pathways construction using tabu search”.
- Yu-Min Lin, Shu-Cherng Fang and Jeffrey L. Thorne.” A tabu search algorithm for maximum parsimony phylogeny inference”.
- Martin Paluszewski, Thomas Hamelryck, Pawel Winter.”Protein Structure Prediction Using Tabu Search and HSE Measure”.
- Qi Shen, Wei-Min Shi and Wei Kong.” Hybrid particle swarm optimization and tabu search approach for selecting genes for tumor classification using gene expression data”.
- Martin Paluszewski, Thomas Hamelryck, and Pawel Winter.” Reconstructing protein structure from solvent exposure using tabu search”.

This work is licensed under a Creative Commons Attribution 4.0 International License.