Chetan R. Dudhagara1, Ashish P. Joshi2, Mayur M. Patel1
1Computer Science Department, N. V. Patel College of Pure And Appllied Sciences – Vallabh Vidyanagar, Gujarat, India.
2BCA Department, VP & RPTP Science College, Vallabh Vidyanagar
Article Publishing History
Article Received on :
Article Accepted on :
Article Published : 21 Jul 2015
Article Metrics
ABSTRACT:
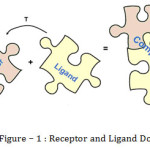
This paper is based on multidisciplinary approach in the fields of computer science (or information technology), bioinformatics and cheminformatics and its effect on modern drug related activities. The computational methods is used to study the formation of inter molecular structures in drug discovery activity has been subject of research during last decades. The drug activity is obtained through molecule binding. The drug activity is obtained through the molecular binding of one molecule (i.e. the ligand) to the pocket of another, usually larger; molecule (i.e. the receptor) Mostly protein is used as a receptor or larger molecule. Cheminformatics is the application of informatics methods to solve problem related to chemicals. The term “cheminformatics” was introduced only few years ago. Each compound is trying to become a stable form. In the area of molecular modeling, molecular docking is the method to predict preferred orientation of one molecule to second molecule when bound to each other to form a stable complex.
KEYWORDS:
Computer Science; Information Technology; Bioinformatics; Cheminformatics; Drug Discovery; Molecular Docking
Copy the following to cite this article:
Dudhagara C. R, Joshi A. P, Patel M. M. Computational Approach in Complex Structure Prediction for Drug Design Activity. Orient.J. Comp. Sci. and Technol;8(2)
|
Copy the following to cite this URL:
Dudhagara C. R, Joshi A. P, Patel M. M. Computational Approach in Complex Structure Prediction for Drug Design Activity. Orient. J. Comp. Sci. and Technol;8(2). Available from: http://www.computerscijournal.org/?p=1954
|
Introcution
In drug design activities, computational methods are used to predict how a particular compound interact with a given protein target. They can be used to assist in building hypotheses about desirable chemical properties when designing the drug and they can be used to refine and modify drug candidates.
Computational Methods can also be used to automate repetitive tasks such as searching large compound databases. Virtual Screening (VS) is a general term for computational methods that use computers to screen a database of virtual drug candidates (called compounds) to identify promising candidates (leads). This can be seen as an alternative to perform laboratory experiments.
The main advantages of computational methods compared to laboratory (wet-lab) experiments are :
- Computational methods are low cost, no any compounds have to be purchased externally or synthesized by a chemist.
- Computational methods is used to investigate various compounds which is not been synthesized yet.
- Huge chemical search space.
Molecular Docking is used in drug design. Most drugs are small molecules, and molecular docking allows the screening of large databases of potential drugs against protein targets
Molecular Docking
The drug design activity obtained through the molecular binding of one molecule i.e. the ligand, to the pocket of another, usually larger, molecule i.e. the receptor, which is commonly a protein. In their binding conformations, the molecules exhibit geometric and chemical complementarily, both of which are essential for successful drug activity. The computational process of searching for a ligand that is able to fit both geometrically and energetically the binding site of a protein is called molecular docking. It is a method that predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex.
Types of molecular docking are:
- Ligands – DNA/RNA Docking
- Ligands – Protein Docking
- Protein – Protein Docking
The main purpose of these types of docking is to predict bound orientation of the complex. It gives insights activity of each molecule at molecular level, sometimes it is difficult to get from experiment techniques also.
Condition for Molecular Docking
A molecule is characterized by a pair i.e. A and B. In this pair A represents a collection of atoms and B represent a collections of bonds between pairs of atoms. Each atom carries standard information, such as its van der waals radius. There are main three types of information are associated with each bond.
The bond length
The distance between atom centers.
The bond angle
The angle between two consecutive bonds.
Rotation
Whether the bond is rotatable or not.
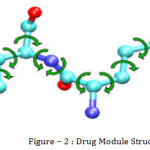
Below figure represent a drug module. In this module, spheres represent atoms. The bonds connecting atoms are represented by sticks. Curve arrows represent the rotatable degrees of freedom around bonds.
Typically each ligands have 3 to 15 rotatable bonds, while each receptors have 1000 to 2000 rotatable bonds. The dimension of the combined searched space makes the docking problem computationally intractable.
Lock and Key Problem
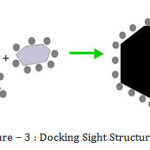
Molecular docking attempts to predict the structure and stability of the complex formed by two interacting molecules, often a protein and its ligand. There may be a problem of “lock-and-key” in docking, where one compound is interested to finding the correct relative orientation of the “key” which will open the “lock”. In this example of docking, the protein can be a “lock” and the ligand can be a “key”. Docking describe the best-fit orientation of a ligand that binds to a particular protein of interest. During docking process, the ligand and the protein adjust conformation to achieve an overall best-fit. The aim of docking is to achieve an optimized conformation for both the ligand and protein and also relative orientation between ligand and protein such that the free energy of overall system is minimized.
Above figure shows the molecular docking process. In this process protein target is represent in black color and a drug candidate or ligand is represent in grey color. In above figure, the curve like structure in the target is the active site upon which the drug attaches. The active site is also called the binding pocket. The active site of protein can be viewed as a lock, and the ligand can be as a key. This figure shows that the molecular docking is the process of testing whether a given key fits a particular lock.
Methods
Search Modes
- To perform a molecular docking, it is required to select and load the ligand and receptor PDB structure which you want to perform a docking.
- It supports three search modes for docking. Mostly in docking used Full Rotation mode, in which the receptor and ligand are effectively rotated on their own centroids, and the ligand is twisted in intermolecular axis.
Docking Example
From the File menu Open Receptor and Ligand.
- From the Control menu select Docking.
- Set various parameters from the Docking Control.
- Perform Fourier Transformation Process, Steric Scan Process, Final Search, Refinement, And Total Dockings Process.
- Representation of Solid Surface
- Representation of Harmonic Surface :
Docking Result
- Mostly, there are two method is used to save molecular docking result in the storage devices.
- The most compact method for save docking result is File …. Save ….
- The second method is to save in separate PDB file. The docking save in to a single PDB file by selecting File…. Save….. Both…
Result and Discussion
Molecular docking is used when the structure of the target element is know. This method is used to test possible hypotheses before conducting the laboratory experiment. Laboratory experiments are comparatively very costly and it takes more time for the experiment. This docking process will try to predict how a drug element is binds with the protein target. This study perform on receptors and ligands in computer without laboratory experiment. This process is very useful for the area of biosciences and chemical sciences for predication of molecule structure and interaction of receptors and ligands.
References
- “Accelerating and Focusing Protein-Protein Docking Correlations Using Multi-Dimensional Rotational FFT Generating Functions” D.W. Ritchie, D. Kozakov, and S. Vajda (2008). Bioinformatics. In Press.
- “Computational Biology and Drug Discovery: From single – network Drugs”, Current Bioinformatics, 2006, 1, 3-13.
- http://www.combichemistry.com/drug-discovery.html
- http://en.wikipedia.org/wiki/Ligand_docking
- Hex 8.0 User Manual.
- New Technology in Drug Discovery. 2003 Powerpoint slides by David J. Wild, Ph.D, University of Michigan.
- “Protein Docking Using Spherical Polar Fourier Correlations”, D.W. Ritchie & G.J.L. Kemp (2000)

This work is licensed under a Creative Commons Attribution 4.0 International License.